DeepMind、分子内の電子の分布を予測するAIを発表
英ロンドンに拠点を置く人工知能(AI)企業DeepMindの研究者を中心とするチームは、分子内の電子の分布を予測することで、その分子の特性を示唆する機械学習モデル「DM21」を開発した。


英ロンドンに拠点を置く人工知能(AI)企業DeepMindの研究者を中心とするチームは、分子内の電子の分布を予測することで、その分子の特性を示唆する機械学習モデル「DM21」を開発した。12月10日発行のサイエンス誌に掲載されたこの手法は、既存の技術よりも正確に一部の分子の特性を算出することができる。
物質や分子の構造は、原理的には量子力学、特に電子の波動関数の振る舞いを支配するシュレーディンガー方程式によって決定される。シュレーディンガー方程式とは、電子の波動関数の振る舞いを規定するもので、空間の特定の位置に特定の電子が存在する確率を表す数学的な小道具だ。
しかし、1960年代にピエール・ホーエンベルグとウォルター・コーンが、それぞれの電子を個別に追跡する必要はないことに気付いた。電子がそれぞれの位置に存在する確率(電子密度)がわかれば、すべての相互作用を正確に計算することができると想定した。このことを証明したコーンはノーベル化学賞を受賞し、密度汎関数理論(DFT)を確立した。
薬理学者からバッテリーエンジニアまで、新しい分子の発見や開発を仕事にしている研究者たちは、何十年もの間、DFTを用いて分子の物理的特性を予測してきた。この理論では、個々の電子をモデル化するのではなく、分子内の電子の負電荷の全体的な分布を計算することを目的としている。
しかし、電子密度と相互作用エネルギーの対応付け、いわゆる密度汎関数の正確な性質は、50年以上にわたって不明であり、近似的に計算するしかなかった。長年にわたり、研究者たちは、精度に差はあるものの、正確な関数の近似式を数多く提案してきた。しかし、これらの近似式はいずれも、正確な関数の重要な数学的特性を捉えていないため、系統誤差を抱えている。
DFTには限界があり、塩化ナトリウムのような単純な分子であっても、塩素原子とナトリウム原子が無限に離れていても、塩素原子がナトリウム原子の電子の何分の1かを保持していることをDFTが誤って予測してしまうことが知られている。また、DFT計算は、基礎的な量子論から始める計算よりもはるかに効率的だが、それでも煩雑で、多くの場合スーパーコンピュータが必要だ。そのため、この10年間で、理論化学者たちは、特に物質の化学反応性や熱伝導性などの特性を調べるために、機械学習を使った実験を行うようになってきた。
今回、DeepMindはニューラルネットワークを用いて、従来よりも正確な電子の密度と相互作用のマップを構築できることを示し、この分野に大きな進歩をもたらした。関数をニューラルネットワークとして表現し、これらの正確な特性を学習データに組み込むことで、重要な系統誤差のない関数を学習し、幅広いクラスの化学反応をよりよく記述することができると研究者は主張している。
研究グループは、小型の主群モデルの特性に関するデータと、これまでの研究で既知のDFTの失敗を解決するのに役立つと考えられていた電子の分数を含む架空の系のデータを用いて、ニューラルネットワークを学習させた。そして、DeepMind21(DM21)と呼ばれるニューラルネットワークを用いて、テスト分子の電子分布を予測した。DM21は、アデニン-チミンのDNA塩基対や水素原子の鎖などのテスト系に対して、標準的なDFTプログラムよりも正確な計算を行った。
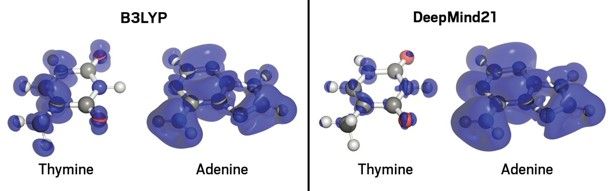
DeepMindのJames Kirkpatrickによると、DFTの標準的な手法は、計算化学者が分子の既知の数学的特性をいくつか取り入れた方程式系を書くというものだそうだ。そのような限られた制約は、特定の状況では、電子共有の例のようなエラーにつながる。一方、ニューラルネットワークは、分子の数学的特性に関する先入観なしに、見たことのあるデータから一連の方程式を補間するだけである。
「DeepMindチームは、DFT計算の最終結果である電子密度の計算にAIを導入するという、おそらくこれまでで最も野心的な試みを行った。これは機械学習にとって理想的な問題のようなものだ」とDFTの研究を長年行ってきた計算化学者で、現在はDeepMindに所属するAlon Cohenは語っている。
DeepMindは学習したシステムを誰でも使えるように公開している。



